$optical-absorption
This keyword allows calculating optical absorption coefficient and solar cells.
$optical-absorption optional destination-directory character required calculate-optics character optional kind-of-absorption character optional read-in-k-points character optional num-quantum-cluster integer optional e-min-state double optional e-max-state double optional e-min-photon double optional e-max-photon double optional num-energy-steps integer optional smoothing-of-curve character optional smoothing-damping-parameter double optional E-P double optional polarization-vector-1 double_array optional polarization-vector-2 double_array optional magnitude-relation-1-2 double optional phase double optional fermi_in_el double optional fermi_in_hl double optional device_thickness_in double optional k-space-symmetry character optional !------------------------------------------------------------- ! The following are only relevant for solar cell simulations. !------------------------------------------------------------- incident-light-along-direction character optional import-absorption-spectrum character optional file-absorption-spectrum character optional import-reflectivity-spectrum character optional file-reflectivity-spectrum character optional import-transmission-spectrum character optional file-transmission-spectrum character optional import-solar-spectrum character optional file-solar-spectrum character optional number-of-suns double optional calculate-black-body-spectrum character optional $end_optical-absorption optional
A tutorial is available that describes this keyword: Optical absorption of an InGaAs quantum well
Detailed description about the Physics: Absorption, Matrix elements, Inter-band transitions, Intra-band transitions (pdf).
- destination-directory
- type:
character
- presence:
required
- example:
optics/
Directory for output of data files.
Optical absorption
- calculate-optics
- type:
character
- options:
yesorno- default:
no
Choose yes if you want to calculate the optical absorption spectrum (Step 3).
This flag can be set to no for Step 1 and Step 2, and yes for Step 3 (see below).
- num-quantum-cluster
- type:
integer >= 1
- default:
1
Number of quantum cluster for which absorption spectrum is calculated.
If this specifier is not present, the quantum cluster 1 is taken.
- kind-of-absorption
- type:
character
- options:
interband-onlyintra-vb-onlyintra-cb-onlyintra-sg-onlyinter-sg-only
interband-onlyConsiders only interband transitions between holes and electrons.
heavy hole
<==>Gamma bandlight hole
<==>Gamma bandsplit-off hole
<==>Gamma band
intra-vb-onlyConsiders only intraband transitions within the valence bands.
heavy hole
<==>light holeheavy hole
<==>split-off holelight hole
<==>split-off holeheavy hole
<==>heavy holelight hole
<==>light holesplit-off hole
<==>split-off hole
intra-cb-onlyConsiders only intraband transitions within the conduction band (Gamma band).
Gamma band
<==>Gamma band
intra-sg-onlyConsiders only intraband transitions within the same band (single-band for Gamma, L, X, heavy hole, light hole, split-off hole band)
Gamma band
<==>Gamma bandL band
<==>L bandX band
<==>X bandheavy hole
<==>heavy holelight hole
<==>light holesplit-off hole
<==>split-off hole
This is a simple algorithm taking only account the energy levels and wave functions at \(k_\parallel=0\) (for single-band case). It only works for 1D and 2D simulations so far. It can also be used for the \(\mathbf{k} \cdot \mathbf{p}\) wave functions as shown in this tutorial: Intersubband transitions in InGaAs/AlInAs multiple quantum well systems. In this case, the correct \(\mathbf{k} \cdot \mathbf{p}\) density and k-dependent matrix elements and nonparabolicity and anisotrpicity of the energy dispersion \(E(k_x,k_y)\) is taken into account.
In order for this flag to work, the following must be present in the input file ($output-1-band-schroedinger):
$output-1-band-schroedinger ... intraband-matrixelements = yes ! or any other value apart from 'no'
The equation used is described here: Intraband absorption in the single-band case
inter-sg-onlySimilar as
intra-sg-onlybut for interband transitions. It currently does not work for \(\mathbf{k} \cdot \mathbf{p}\) wave functions.
Specifications for energy range of absorption spectrum: lower/upper boundary for photon energy interval
- e-min-photon
- type:
double
- unit:
(eV)- example:
1.0
lower boundary for photon energy
- e-max-photon
- type:
double
- unit:
(eV)- example:
2.0
upper boundary for photon energy
- num-energy-steps
- type:
integer
- example:
1000
Number of energy steps between e-min-photon and e-max-photon.
This number determines the resolution of the absorption spectrum curve \(\alpha(E)\)
where \(E\) is the energy in units of (eV).
Note
The number of
energy grid points =
num-energy-steps\(+1\)
because the first grid point (e-min-photon) is also included.
The energy grid spacing is
\(\Delta_E = (\)
e-max-photon\(-\)e-max-photon\() /\)num-energy-steps.
Distinguish between calculating and reading in \(k_\parallel\) points.
- read-in-k-points
- type:
character
- options:
yesorno- default:
no
Flag to distinguish between Step 2 and Step 3. For Step 3, in order to avoid calculating the \(k_\parallel\) points again, one can simply read them in from a previous simulation, and then calculate and output the optical absorption spectrum.
Energy of lowest/highest eigenvalue considered for calculation, i.e. energies are calculated in the interval [e-min-state,e-max-state].
Here, the eigenvalue solver is called with these energy values.
Alternatively, the eigenvalue solver can be called with a certain number of eigenvalues requested, i.e. one either has to specify an energy interval or the number of eigenvalues.
Depending on the eigenvalue solver used, different options are possible.
- e-min-state
- type:
double
- unit:
(eV)- default:
-5.0
- example:
-1.7
lowest eigenvalue
- e-max-state
- type:
double
- unit:
(eV)- default:
5.0
- example:
0.3
highest eigenvalue
Broadening of absorption curve
This is only relevant for the calculation of the absorption spectrum.
- smoothing-of-curve
- type:
character
- options:
Lorentzian,Gaussian,yes,no- default:
yes
For Lorentzian or Gaussian, we introduce an artificial broadening (smoothing) of the curve (Lorentzian or Gaussian broadening).
If yes, both Lorentzian and Gaussian broadening will be calculated and written out.
If no, no broadening (smoothing) of the curve is assumed.
- smoothing-damping-parameter
- type:
double > 0.0
- unit:
(eV)- default:
0.005
The artificial parameter for smoothing of absorption spectrum is smoothing-damping-parameter.
It is usually denoted as \(\Gamma\) and is the Full Width at Half Maximum (FWHM).
Lorentzian lineshape
The Lorentzian function is given by
\(L(E) = \frac{1}{\pi} \frac{\Gamma/2}{(E-E_{ij})^2+(\Gamma /2)^2}\)
where \(\Gamma /2\) is the scale parameter Lorentzian half-width, i.e. Half Width at Half Maximum (HWHM). It describes the shape of certain types of spectral lines (lineshape). Note that the definition of the Lorentzian function includes a factor \(1/\pi\).
\(E_{ij}\) is the transition energy between the states \(i\) and \(j\) and specifies the location of the peak in the Lorentzian function.
\(\Gamma\) is specified in the input file via
smoothing-damping-parameter. It is the Full Width at Half Maximum (FWHM).\(\alpha=\Gamma /2\) is the Half Width at Half Maximum (HWHM).
!-------------------------------------------- ! for k.p algorithm only: ! First, the absorption spectrum is calculated. ! Then the broadening is applied. !-------------------------------------------- absorption_NoSmoothingV = absorptionV absorptionV = 0.0 DO i=1,num-energy-steps+1 ! Loop over all energy grid points E(i) and determine absorption coefficients alpha(i)=alpha(E). DO j=1,num-energy-steps+1 ! This loop is essentially an integration over energy dE. E_weight = (Lorentzian( E_gridV(j), E_gridV(i), smoothing-damping-parameter ) - & Lorentzian( E_gridV(j), - E_gridV(i), smoothing-damping-parameter ) ) * DeltaEnergy absorptionV(i) = absorptionV(i) + absorption_NoSmoothingV(j) * E_weight END DO END DO
The following specifiers are only used for the \(\mathbf{k} \cdot \mathbf{p}\) optical absorption spectrum but not for the simple single-band intersubband absorption algorithm.
- E_P
- type:
double
- example:
20.0- unit:
(eV)- status:
currently not implemented, value from the database is used
\(E_P\) is Kane’s matrix element \(E_P = | < S | p | X > |^2\).
It should be around 20 eV and depends on the material.
The E_P parameter is given in the database by the specifier 8x8kp-parameters.
\(E_P\) can be converted into the \(P\) paramter by the following equation:
\(E_P = \frac{2 m_0}{\hbar^2} P^2\).
In our model the \(E_P\) parameter is only relevant for interband transitions.
It enters into the matrix element prefector (matrix_element_prefac) which is described in
Section 1.1.1 Inter-band transitions of the documentation:
Absorption spectrum, Matrix elements, Inter-band transitions, Intra-band transitions (pdf).
\(E_P\) has the same value for all materials in this implementation.
In principle it could have been read in from the database rather than specifying it within the keyword $optical-absorption.
Polarization
\(m\) is equivalent to \(\tan(\theta)\) or \(\tan^{-1}(\theta)\) depending whether \(P_1/P_2\) is \(x\) or \(y\). so instead of \(\theta\) one can directly use the formula below:
\(P = m \cdot P_1 + e^{\text{i}\omega \pi} \cdot P_2\)
This is in fact more general, as it also describes circular polarization which lead to complex coefficient.
- polarization-vector-1
- type:
double array
- example:
1.0 0.0 0.0
x y z coordinates (in simulation system) for first in-plane vector \(P_1\)
- polarization-vector-2
- type:
double array
- example:
0.0 1.0 0.0
x y z coordinates (in simulation system) for second in-plane vector \(P_2\)
- magnitude-relation-1-2
- type:
double
- default:
0.5- example:
1.0
relation of magnitudes \(m=|E_1|/|E_2|\)
- phase
- type:
double
- default:
0.0- example:
0.5
phase \(\omega\): \(E_2\) ==> \(\exp(\text{i} \omega \pi)E_2\)
\(P = m \cdot P_1 + e^{\text{i}\omega \pi} \cdot P_2\)
Examples
x-polarized light
polarization-vector-1 = 0.0 1.0 0.0 polarization-vector-2 = 1.0 0.0 0.0 magnitude-relation-1-2 = 0.0In this case,
polarization-vector-1is ignored as \(|E_1|\) is set to be zero.z-polarized light
polarization-vector-1 = 1.0 0.0 0.0 polarization-vector-2 = 0.0 0.0 1.0 magnitude-relation-1-2 = 0.0In this case,
polarization-vector-1is ignored as \(|E_1|\) is set to be zero.circularly polarized light in the (x,y) plane
polarization-vector-1 = 1.0 0.0 0.0 polarization-vector-2 = 0.0 0.0 1.0 magnitude-relation-1-2 = 1.0In this case,
polarization-vector-1is not ignored as \(|E_1|=|E_2|\).quantum well, interband absorption
polarization-vector-1 = 1.0 0.0 0.0 polarization-vector-2 = 0.0 1.0 0.0 magnitude-relation-1-2 = 1.0quantum well, intraband absorption
polarization-vector-1 = 0.0 0.0 1.0 polarization-vector-2 = 0.0 0.0 1.0 magnitude-relation-1-2 = 0.0Note: Intraband absorption spectrum only for z-polarized light.
Fermi levels
- fermi_in_el
- type:
double
- unit:
(eV)- default:
0.0- example:
0.1
Optional input for Fermi level of electrons (default: calculated quasi-Fermi level for electrons)
- fermi_in_hl
- type:
double
- unit:
(eV)- default:
0.0- example:
-1.0
Optional input for Fermi level of holes (default: calculated quasi-Fermi level for holes)
- device-thickness
- type:
double
- unit:
(m)- default:
thickness of device or quantum cluster? (Check this!)
- example:
1e-6
Optional input of device thickness for normalization of absorption spectrum
- k-space-symmetry
- type:
character
- options:
default,none,four-fold- default:
default
Symmetry of \(k_\parallel = (k_x,k_y)\) space to be discretized. If any symmetry is present, less \(k_\parallel\) points have to be calculated. By default, the appropriate symmetry is chosen taking into account any crystal rotations with respect to the simulation axes, as well as nonsymmetric strains.
Note
In order to save CPU time, we recommend the following procedure:
Instead of calculating
\(\mathbf{k} \cdot \mathbf{p}\) eigenstates and
optical absorption spectrum
within one simulation, it is more efficient to divide this into 3 steps.
Step 1: Calculate eigenstates for \(k_\parallel=0\).
calculate-optics = no
This is very quick. Solve \(\mathbf{k} \cdot \mathbf{p}\) to determine lowest and highest eigenvalue so that we know what to specify for
e-min-stateande-max-statein Step 2.Step 2: Calculate eigenstates for all \(k_\parallel\) vectors and save all of the wave functions to file so that they can be read in and used many times in Step 3.
raw-potential-in = yes raw-fermi-levels-in = yes strain-calculation = raw-strain-in num-kp-parallel = 1700 ! STEP 2/3 ! total number of k_|| points for Brillouin zone discretization calculate-optics = yes num-quantum-cluster = 1 read-in-k-points = no e-min-state = -1.7 ! Choose a reasonable value for E_min e-max-state = 0.3 ! Choose a reasonable value for E_min
Read in raw data (potential, quasi-Fermi levels, strain (if applicable) and all \(\mathbf{k} \cdot \mathbf{p}\) wave functions) and output \(k_\parallel\) points. The strain calculation is very fast for a 1D simulation. Instead of reading it in, it could be directly calculated (recommended).
The user specifies the total number of \(k_\parallel\) points that are present in \(k_\parallel\) space. However, internally the code modifies this number according to the following algorithm:
number of k points in positive x direction (without Gamma point): N_kx
number of k points in positive y direction (without Gamma point): N_ky = N_kx
==>Thus the actual, total number of \(k_\parallel\) points is:total_number_of_k|| = (2 * N_kx + 1) * (2 * N_ky + 1)In this example (
num-kp-parallel = 1700):N_kx = N_ky = 20==> total_number_of_k|| = 41 * 41 = 1681Step 3: Calculate optical absorption spectrum.
calculate-optics = yes read-in-k-points = yes
Read in \(k_\parallel\) points, calculate and output optical absorption spectrum for specific polarization of incident light.
If one wants to repeat the calculation for another polarization, one only needs to change the polarization vector and repeat Step 3. It is not necessary in this case to recalculate Step 1 or Step 2. Step 3 also outputs the energy dispersion \(E(k_\parallel)= E(k_x,k_y)\). (Check: Why not Step 2?)
Output
Results
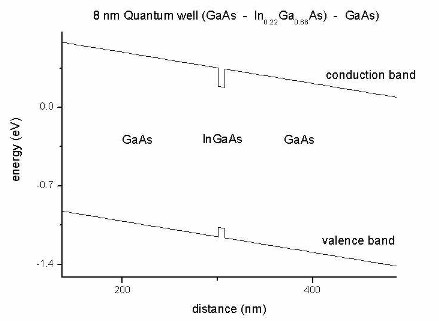
Figure 2.3.1 Conduction and valence band edges of the quantum well
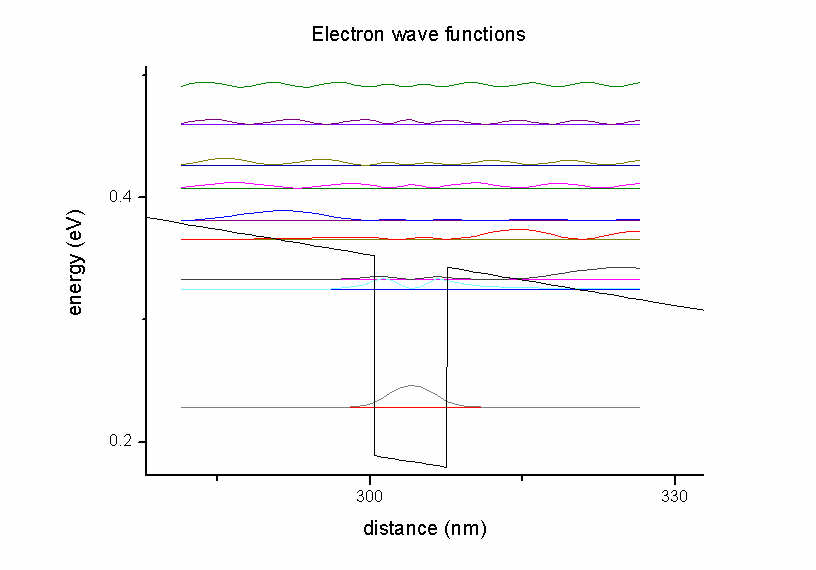
Figure 2.3.2 Electron states in the quantum well
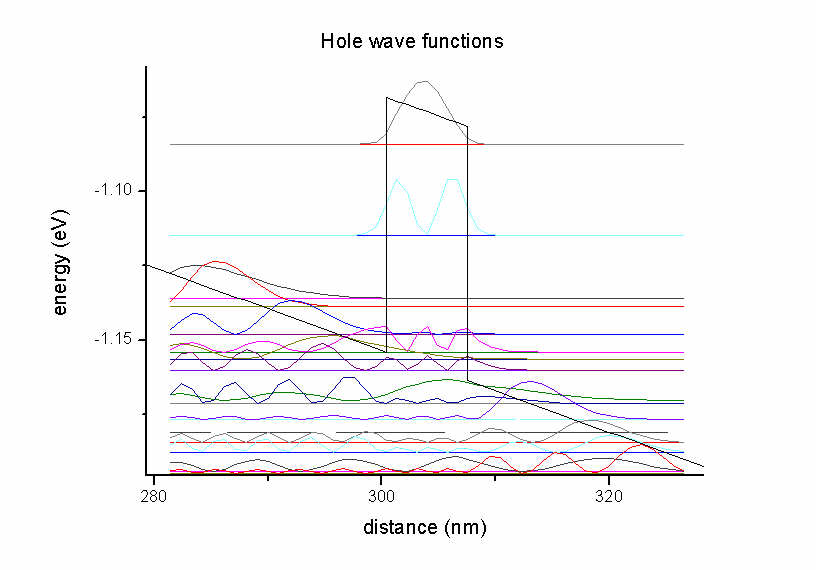
Figure 2.3.3 Hole states in the quantum well
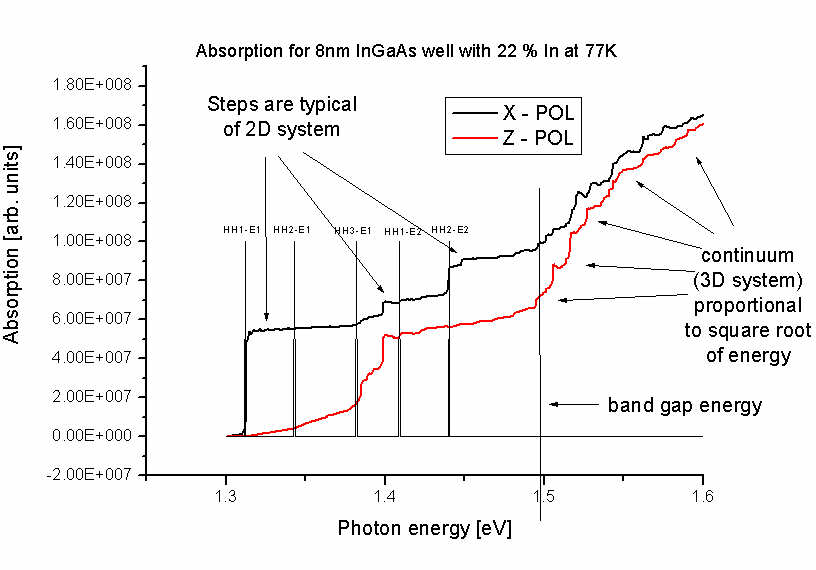
Figure 2.3.4 Optical absorption spectrum of the quantum well
The unit of the optical absorption coefficient is (m^-1) and not arbitrary units as indicated in the figure.
The electric susceptibility tensor \(\chi\) is contained in the file susceptibility_tensor.dat:
chi11re chi11im chi22re chi22im chi33re chi33im chi12re chi12im chi13re chi13im chi23re chi23im
Note: As this tensor is complex, for each component, two values are written out.
re: real partim: imaginary part
The relevant part for the absorption spectrum is only the imaginary part.
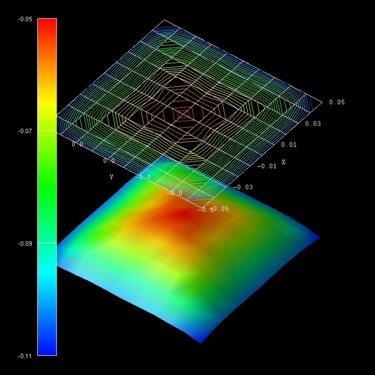
Figure 2.3.5 Energy dispersion \(E(k_x,k_y)\) of the highest hole eigenstate (ground state).
The units of the \(k_\parallel\) space grid coordinates \(k_x\) and \(k_y\) are (Angstrom^-1) and the energy units are (eV).
The files
el_dispersion_100.datel_dispersion_110.dathl_dispersion_100.dathl_dispersion_110.dat
show the same data but with slices along the
[10] (i.e. \(k_\parallel=(k_x,k_y=0)\) and
[11] (i.e. \(k_\parallel=(k_x=k_y)\) directions in \(k_\parallel\) space.
Here all electron and all hole eigenvalues are contained in one file, respectively.
Restrictions
Only Dirichlet boundary conditions are supported so far.
Step 2 and Step 3 only work if:
raw-potential-in = yes
Solar cells
For solar cells, we have this tutorial: GaAs Solar Cell
Example files for solar spectra, absorption coefficient, transmission and reflectivity coefficients can be found in the installation folder:
C:\Program Files\nextnano\nextnano3\Syntax\Solar cell files\absorption\C:\Program Files\nextnano\nextnano3\Syntax\Solar cell files\reflectivityC:\Program Files\nextnano\nextnano3\Syntax\Solar cell files\solar spectra\
The following specifiers are relevant for solar cell simulations (photovoltaics).
- incident-light-along-direction
- type:
character
- options:
x,y,z,-x,-y,-z- default:
along simulation direction in 1D
In a 1D simulation, this specifier is optional. For 2D and 3D, a direction must be specified.
Solar spectrum
- import-solar-spectrum
- type:
character
- options:
yesorno- default:
no
For a solar cell simulation, one has to read in a solar spectrum, e.g. AM 1.5, or AM 1.0 (AM = air mass). They can be obtained from NREL website, e.g. ASTM-E490: https://www.nrel.gov/grid/solar-resource/spectra-astm-e490.html (AMST = American Society for Testing and Materials)
- file-solar-spectrum
- type:
character
- example:
H:\solar_cells\ASTMG173_AM10.datAM 1.0 spectrum (extraterrestrial)- example:
H:\solar_cells\ASTMG173_AM15.datAM 1.5 spectrum- example:
H:\solar_cells\ASTMG173_AM15G.datAM 1.5G spectrum (G = global, i.e. including diffuse light)
The file must consist of two columns (wavelength and spectrum), the units are (nm) and (W/m^2*nm^-1).
wavelength[nm] AM1.5[W/m^2*nm^-1] ... ...
Concentration of sun light
- number-of-suns
- type:
integer
- default:
1.0our sun- example:
0.0(no sun, dark)- example:
2.52.5 suns- example:
300.0300 suns- example:
1000.0100 suns
The number of suns can be set to increase the power of the solar spectrum in order to model concentrator solar cells.
Absorption Spectra
- import-absorption-spectrum
- type:
character
- options:
yesorno- default:
no- file-absorption-spectrum
- type:
character
- example:
AbsorptionCoefficient_GaAs_300K.dat
The file must consist of two columns (wavelength and absorption coefficient), the units are (nm) and (cm^-1).
wavelength[nm] absorption[1/cm] ... ...
Reflection coefficient
Fraction of incident photons that are reflected from surface for a particular wavelength.
- import-reflectivity-spectrum
- type:
character
- options:
yesorno- default:
no- file-reflectivity-spectrum
- type:
character
- example:
ReflectionCoefficient_GaAs_300K.dat
The file must consist of two columns, wavelength (nm) and reflection coefficient.
wavelength[nm] reflectivity[] ... ...
Transmission coefficient
Fraction of incident photons that are transmitted through the device for a particular wavelength (relevant for very thin devices).
- import-transmission-spectrum
- type:
character
- options:
yesorno- default:
no- file-transmission-spectrum
- type:
character
- example:
TransmissionCoefficient.dat
The file must consist of two columns, wavelength (nm) and transmission coefficient.
wavelength[nm] transmission[] ... ...
Solar cell output
All output is twofold:
one is with respect to wavelength in units of
(nm)one is with respect to photon energy in units of
(eV)(indicated by_eV*.dat)
The files are:
Absorption coefficient
optics/Absorption_coefficient.dat(as read in from file but now in units of(m^-1))optics/Absorption_coefficient_interpolated.dat(interpolated on wavelength grid of solar spectrum but now in units of(m^-1))
Reflectivity
optics/Reflectivity.dat(as read in from file)optics/Reflectivity_interpolated.dat(interpolated on wavelength grid of solar spectrum)
Transmission
optics/Transmission.dat(as read in from file)optics/Transmission_interpolated.dat(interpolated on wavelength grid of solar spectrum)
Solar spectrum
optics/SolarSpectralIrradiance_sun0001.dat(as read in from file)optics/PhotonFlux_sun0001.dat(photon flux density calculated from solar spectrum)
Total number of of photons in the solar spectrum above an energy value contributing to the maximum photocurrent for a solar cell made with a specific band gap:
optics/PhotonFlux_BandGap_eV_sun0001.dat(calculated from solar spectrum)optics/PhotoCurrent_BandGap_eV_sun0001.dat(calculated from solar spectrum)
Spectral response
optics/SpectralResponse_sun0001.datexternal and internal spectral response
Quantum efficiency
optics/QuantumEfficiency_sun0001.datexternal and internal quantum efficiency
Generation rate
optics/GenerationRateLight_AVS_sun0001.fld2D plots \(G(x,\lambda)\) and \(G(x,E)\)optics/GenerationRateLight_sun0001.dat1D plot \(G(x)\)optics/GenerationRate_eV_sun0001.dat1D plot \(G(E)\) where \(E\) is the energyoptics/GenerationRate_Wavelength_sun0001.dat1D plot \(G(\lambda)\)
Current-voltage characteristics
current/IV_characteristics_new.datvoltage[V] current[A/m^2] ... power[W/m^2] powersolar[W/m^2] efficiency[%]
The following information can be found in the
.logfile, such asshort-circuit current \(I_\text{sc}\)
open-circuit voltage \(U_\text{oc}\)
ideal conversion efficiency \(\eta\)
…
**************************************************************************************** Solar cell results **************************************************************************************** short-circuit current: I_sc = 281.473346 [A/m^2] (photo current: It increases with smaller band gap.) open-circuit voltage: U_oc = -1.012500 [V] (U_oc <= built-in potential ~ band gap) current at maximum power: I_max = 273.089897 [A/m^2] voltage at maximum power: U_max = -0.925000 [V] maximum power output: P_max = U_max * I_max = -252.608155 [W/m^2] (condition for maximum power output: dP/dV = 0) maximum extracted power: P_solar = - P_max = 252.608155 [W/m^2] incident power: P_in = 0.000000 [W/m^2] ideal conversion efficiency: eta = P_max / P_in = Infinity % fill factor: FF = 0.886370 In practice, a good fill factor is around 0.8. All these results are approximations. They are only correct if a lot of voltage steps have been used (i.e. a high resolution). ****************************************************************************************
Example for a solar cell simulation
!-------------------------------------- $optical-absorption destination-directory = optics/ import-absorption-spectrum = yes file-absorption-spectrum = "..\Syntax\Solar cell files\absorption\AbsorptionCoefficient_GaAs_300K.dat" import-reflectivity-spectrum = yes file-reflectivity-spectrum = "..\Syntax\Solar cell files\reflectivity\Reflectivity_Al0.80Ga0.20As.dat" import-solar-spectrum = yes file-solar-spectrum = "..\Syntax\Solar cell files\solar spectra\ASTMG173_AM15G.dat" number-of-suns = 1 $end_optical-absorption !--------------------------------------
Black body spectrum
- calculate-black-body-spectrum
- type:
character
- options:
yesorno- default:
no
Flag for calculating black body spectrum according to Planck’s law, e.g. to compare the solar spectrum to the spectrum of a black body at T = 5778 K.
{kind=link}
The spectral energy density
the spectral radiance (which is emitted per m2 and per unit solid angle sr (sr = steradian)) and
the spectral irradiance (which is received per m2)
is calculated.
Note
spectral irradiance = spectral radiance \(\cdot \pi\)
spectral energy density = spectral radiance \(\cdot 4\pi/c\)
There are several output files, i.e. output with respect to
wavelength \(\lambda\) in units of
(m),BlackBody_SpectralEnergyDensity_wavelength.datWavelength[nm] SpectralEnergyDensity[kJ/m^3/m]BlackBody_SpectralRadiance_wavelength.datWavelength[nm] SpectralRadiance[kW/m^2/nm/sr]BlackBody_SpectralIrradiance_wavelength.datWavelength[nm] SpectralIrradiance[kW/m^2/nm]
angular frequency \(\omega = 2 \pi \nu\) in units of
(1/s),BlackBody_SpectralEnergyDensity_angular_frequency.datAngularFrequency_omega[10^15/s] SpectralEnergyDensity[10^-15J/m^3/s^-1]BlackBody_SpectralRadiance_angular_frequency.datAngularFrequency_omega[10^15/s] SpectralRadiance[10^-12W/m^2/s^-1/sr]BlackBody_SpectralIrradiance_angular_frequency.datAngularFrequency_omega[10^15/s] SpectralIrradiance[10^-12W/m^2/s^-1]
frequency \(\nu\) in units of
(Hz),BlackBody_SpectralEnergyDensity_frequency.datFrequency[THz] SpectralEnergyDensity[10^-15J/m^3/Hz]BlackBody_SpectralRadiance_frequency.datFrequency[THz] SpectralRadiance[10^-12W/m^2/sr]BlackBody_SpectralIrradiance_frequency.datFrequency[THz] SpectralIrradiance[10^-12W/m^2/Hz]
photon energy \(E = h \nu\) in units of
(eV).BlackBody_SpectralEnergyDensity_energy.datAngularFrequency_omega[10^15/s] SpectralEnergyDensity[kJ/m^3/eV]BlackBody_SpectralRadiance_energy.datAngularFrequency_omega[10^15/s] SpectralRadiance[kW/m^2/eV/sr]BlackBody_SpectralIrradiance_energy.datAngularFrequency_omega[10^15/s] SpectralIrradiance[kW/m^2/eV]
The file BlackBody_Info.txt contains some additional information about the calculated black body spectrum.
Intraband absorption in the single-band case
In the following we assume a single band with a parabolic energy band dispersion.
Tutorials showing results are available here:
For a 1D heterostructure grown along the \(x\) direction, formula for the absorption coefficient \(\alpha\) reads (see e.g. [ChuangOpto1995] or p. 53 in [FaistQCL2013])
\(\alpha(\omega) = \frac{e^2 \omega}{\varepsilon _0 n_\text{r}c} \sum_{i} \sum_{j} \left( \overline{n}_i - \overline{n}_j\right) x_{ij}^2 \frac{\Gamma/2}{\left(E_j-E_i-\hbar\omega\right)^2+(\Gamma /2)^2}\)
or, equivalently in energy,
\(\alpha(E) = \frac{e^2 E}{\hbar \varepsilon _0 n_\text{r}c} \sum_{i} \sum_{j} \left( \overline{n}_i - \overline{n}_j\right) x_{ij}^2 \frac{\Gamma/2}{\left(E_j-E_i-\hbar\omega\right)^2+(\Gamma /2)^2}\)
where
\(\omega=E/\hbar\) is the frequency in units of
(s^-1)\(E\) the energy in units of
(J)\(e\) is the elementary charge in units of
(As)\(\varepsilon _0\) is the vacuum permittivity in units of
(As/Vm)\(c\) is speed of light in vacuum in units of
(m/s)\(n_\text{r} = \sqrt{ \varepsilon _\text{r} }\) is the refractive index assumed to be homogeneous. So we take the average of the quantum region (check this).
\(\overline{n}_i=\frac{1}{L}\sigma _i=\frac{1}{L} \int n_i(x) \text{d}x\) is the averaged electron density of subband \(i\) in units of
(m^-3), where \(L\) is the length of the quantum region and \(\sigma _i=\int n_i(x) \text{d}x\) is the subband density in units of(m^-2)\(x_{ij}=<i|x|j>\) is the dipole moment between initial state \(i\) and final state \(j\) in units of
(m)\(\Gamma\) is the energy linewidth (broadening) in units of
(J)in terms of full-width at half maximum (FWHM).
This equation includes a Lorentzian broadening which includes a factor of \(1/\pi\).
We can also define the position dependent absorption coefficient
\(\alpha(\omega,x) = \frac{e^2 \omega}{\varepsilon _0 n_\text{r}c} \sum_{i} \sum_{j} \left( n_i(x) - n_j(x)\right) x_{ij}^2 \frac{\Gamma/2}{\left(E_j-E_i-\hbar\omega\right)^2+(\Gamma /2)^2}\)
where
\(n_i(x)\) is the electron density of state \(i\) at position \(x\) in units of
(m^-3).
The units of both \(\alpha(\omega)\) and \(\alpha(\omega,x)\) are (m^-1).
In plots, typically (cm^-1) is used.
If we integrate \(\alpha(\omega,x)\) over position \(x\) in the whole quantum region of length \(L\), and divide by the length of the quantum region \(L\), we obtain \(\alpha(\omega)\) as defined above,
\(\alpha(\omega) = \frac{1}{L} \int \alpha(\omega,x) \text{d}x\).
So \(\alpha (\omega)\) as defined in the beginning of this section, where we averaged the density \(\overline{n}_i\), is the averaged absorption coefficient in the quantum region and equivalent to the definition given here.
Finally, we note that this also works for the \(\mathbf{k} \cdot \mathbf{p}\) wave functions:
\(\alpha(\omega,x,k_{\parallel}) = \frac{e^2 \omega}{\varepsilon _0 n_\text{r}c} \sum_{i} \sum_{j} \sum_{k{\parallel}} \left( n_i(x,k_{\parallel}) - n_j(x,k_{\parallel})\right) (x_{ij}(k_{\parallel}))^2 \frac{\Gamma/2}{\left(E_j(k_{\parallel})-E_i(k_{\parallel})-\hbar\omega\right)^2+(\Gamma /2)^2}\)
where
\(n_i(x,k_{\parallel})\) is the electron density of state \(i\) at position \(x\) and vector \(k_{\parallel}=(k_x,k_y)\) (in 1D) or \(k_{\parallel}=k_z\) (in 2D).